1) Proteinuria masiva, con pérdida diaria de proteínas en orina de 3,5 g o más, en adultos
2) Hipoalbuminemia con, concentraciones de albúmina plasmática inferiores a 3 g/dl
3) Edema generalizado, la manifestación clínica más obvia, y
4) hiperlipidemia y lipiduria.
Al principio hay poca o ninguna azotemia, hematuria o hipertensión.
Los componentes del síndrome nefrórico tienen una relación lógica uno con otro. El acontecimiento inicial es un deterioro en las paredes capilares del glomérulo, dando lugar a un aumento de la permeabilidad para proteínas plasmáticas. Debe recordarse, que la pared de los capilares glomerulares, con su endotelio, MBG y podocitos, actúa como una barrera a través de la que debe pasar el filtrado glomerular. Cualquier aumento de permeabilidad resultante de alteraciones estructurales o fisicoquímicas permite que escapen proteínas desde el plasma hasta el filtrado glomerular. Ante una proteinuria de larga duración o extremadamente alta, la albúmina sérica está disminuida, dando lugar a hipoalbuminemia. El edema generalizado del síndrome nefrótico es, a su vez, consecuencia de la caída en la presión osmótica coloidal del plasma como resultado de la hipoalbuminemia y la retención primaria de sal y agua por el riñón. A medida que el líquido escapa del árbol vascular a los tejidos, hay una caída concomitante en el volumen plasmático con filtración glomerular disminuida. La secreción compensatoria de aldosterona, junto con la reducción del FG y la reducción de secreción de péptidos natriuréticos, promueve la retención de sal y agua por los riñones, agravando así aún más el edema. Por la repetición de esta cadena de acontecimientos, puede desarrollarse un edema generalizado (denominado anasarca). La hipoalbuminemia desencadena una síntesis aumentada de lipoproteínas en el hígado. Asimismo, hay un transporte anormal de partículas lipídicas circulantes y deterioro de la fragmentación periférica de las lipoproteínas. A su vez, la lipiduria refleja el aumento de permeabilidad de la MBG para lipoproteínas.
Las lesiones glomerulares primarias más importantes que llevan característicamente al síndrome nefrótico son glomerulosclerosis focal y segmentaria (GS FS) y la enfermedad de cambios mínimos (ECM). Esta última es más importante en niños, y la primera lo es en adultos. Otras dos lesiones primarias, la nefropatía membranosa y la GN membranoproliferativa, también producen síndrome nefrótico.

Enfermedad de cambios mínimos:
Este trastorno, relativamente benigno, es la causa más frecuente de síndrome nefrótico en niños. Se caracteriza por glomérulos con apariencia normal al microscopio óptico pero muestra un borrado difuso de los pedicelios de los podocitos cuando se observan con el microscopio electrónico. Aunque puede desarrollarse a cualquier edad, esta afección es más común entre 1 y 7 años de edad.
Patogenia: Sobre la base de algunos estudios experimentales, se ha atribuido la proteinuria a un factor derivado del linfocito T que produce daño en el podocito y desprendimiento de sus pedicelios. Sin embargo, no se ha establecido la naturaleza de tal factor hipotético ni una función causal de los linfocitos T en la enfermedad humana, y no hay un buen modelo experimental de la enfermedad de cambios mínimos.
Morfologia: Al microscopio óptico, los glomérulos en la enfermedad de cambios mínimos parecen normales. Las células de los túbulos contorneados proximales estan densamente cargados de lípidos y gotitas proteicas con frecuencia, pero esto es secundario a la reabsorción tubular de las lipoproteínas vertidas por los glomérulos enfermos. El aspecto de los túbulos contorneados proximales es la base de la denominación antigua de esta afección, nefrosis lipoide. Incluso con el microscopio electrónico, la MBG parece normal. La única anormalidad glomerular obvia es el borrado uniforme y difuso de los pedicelios de los podocitos. Así pues el citoplasma de los podocitos parece aplanado en los extremos de la MBG, obliterando el entramado de arcos entre los podocitos y la MBG. Asi mismo, hay una vacuolización de la célula epitelial, formación de microvellosidades y desprendimientos focales ocasionales. Cuando los cambios en los podocitos revierten ( p ej., en respuesta a los corticosteroides), la proteinuria remite.

 |
| A) Lesiones mínimas con presencia de lipófagos en intersticio (PAS, 100×). B) Estudio ultraestructural: fusión pedicular continua y aspecto vellositario de los podocitos. |
Evolución clínica: La enfermedad se manifiesta por una evolución insidiosa de síndrome nefrótico en un niño por lo demás sano. No hay hipertensión, y la función renal está conservada en la mayoría de los individuos. La pérdida de proteínas habitualmente está limitada a las proteínas séricas más pequeñas, principalmente la albúmina (proteinuria selectiva). El pronóstico en niños que tienen esta afección es bueno. Más del 90% de los casos responde a un curso breve de corticoterapia; sin embargo, la proteinuria recidiva en más de dos terceras partes de los que responden inicialmente, algunos de los cuales se hacen dependientes de los corticoides. Menos del 5% desarrollan insuficiencia renal crónica después de 25 años, y es probable que la mayoría de las personas de este subgrupo tenga un síndrome nefrótico producido por glomerulosclerosis focal y segmentaria no detectada por biopsia. Dada su respuesta a la terapia en niños, la enfermedad de cambios mínimos debe diferenciarse de otras causas de síndrome nefrótico en los que no responden. Los adultos con enfermedad de cambios mínimos responden, asimismo, a terapia esteroidea, pero la respuesta es más lenta y las recidivas, más habituales.
Glomeruloesclerosis focal y segmentaria:
La GSFS es una lesión caracterizada histológicamente por esclerosis que afecta a algunos, pero no a todos, los glomérulos (afectación focal) y sólo a algunos segmentos de cada glomérulo afectado. Este cuadro histológico se asocia frecuentemente a síndrome nefrórico y puede darse: 1) asociado con otras afecciones conocidas, como infección por el virus de la inmunodeficiencia humana o consumo de heroína (nefroparía por virus de la inmunodeficiencia humana, nefroparía por heroína); 2) como un acontecimiento secundario a otras formas de GN (p.ej ., nefropatía por inmunoglobulina A [lgA]); 3) como una maladaptación tras la pérdida de nefronas; 4) en formas heredadas o congénitas que son el resultado de mutaciones que afectan a proteínas citoesqueléticas o relacionadas que se expresan en los podocitos (p. ej . nefrina ), o 5) como una enfermedad primaria.
La GSFS primaria (o idiopática) es responsable del 20 al 30%, aproximadamente, de todos los casos del síndrome nefrórico. Cada vez es una causa más habitual de síndrome nefrórico en adultos y sigue siendo una causa frecuente en niños. En los niños es importante distinguir esta causa de sindrome nefrótico de la ECM, dado que los cursos clínicos son marcadamente diferentes.
Patogenia: Se desconoce la patogenia de la GSFS primaria. Algunos investigadores han sugerido que la GSFS y la ECM son parte de un todo continuo y que la ECM puede transformarse en GSFS. Otros creen que ambas son distintas entidades clinicopatológicas. En cualquier caso, se cree que la lesión de los podocitos representa el acontecimiento inicial de la GSFS primaria. Lo mismo que en la ECM, se han propuesto factores que aumentan la permeabilidad producidos por los linfocitos. El depósito de masas hialinas en los glomérulos representa el atrapamiento de proteínas plasmáticas y lípidos en los focos de lesión donde se desarrolla la esclerosis. Asimismo, se cree que la IgM y las proteínas del complemento que se observan habitualmente en las lesiones son el resultado del atrapamiento inespecífico por los glomérulos dañados. La recidiva de proteinuria en algunas personas con GSFS que reciben injertos renales, a veces ya a las 24 horas del trasplante, apoya la idea de que la causa es un mediador circulante que daña los podocitos.
Morfologia: En la GSFS, la enfermedad afecta primero solamente a algunos de los glomérulos (de aquí el término focal) e inicialmente sólo a los glomérulos yuxtamedulares. Al progresar, se afectan todos los niveles de la corteza. Histológicamente, la GSFS se caracteriza por lesiones que ocurren en algunos penachos dentro del glomérulo y respetan los otros (de aquí el término segmentarío). Así pues, la implicación es focal y también segmentaria. Los glomérulos afectados muestran aumento de la matriz mesangial, luces capilares obliteradas y depósito de masas hialinas (híalínosis) y gotitas de lípidos. Ocasionalmente, los glomérulos están completamente esclerosados (esclerosis global). En los glomérulos afectados, la microscopia de fluorescencia revela con frecuencia atrapamiento inespecífico de inmunoglobulinas, usualmente IgM, y complemento en las zonas de hialinosis. En microscopía electrónica, los podocitos muestran borrado de los pedicelos, lo mismo que en la ECM. Con el tiempo, la progresión de la enfermedad lleva a la esclerosis global de los glomérulos con atrofia tubular pronunciada y fibrosis intersticial.
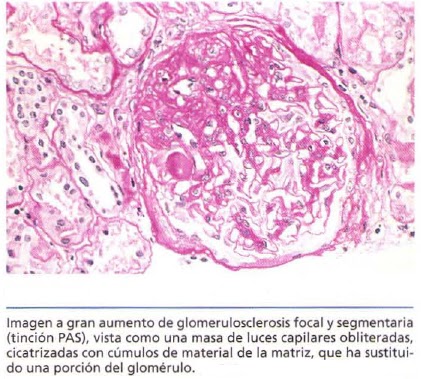
 |
| A) Esclerosis focal segmentaria con «corona» de los pedicelos (PAS, 200×). B) Microscopia electrónica de área glomerular con fusión pedicular, incremento de matriz mesangial y depósitos electrodensos. |
Evolución clínica: Existe poca tendencia a la remisión espontánea en la GSFS idiopática, y habitualmenre la respuesta a la corticoterapia es mala. La progresión a insuficiencia renal ocurre a diferentes velocidades, y aproximadamente el 50% de los individuos tiene insuficiencia renal a los 10 años.
Nefropatía membranosa ( glomerulonefritis membranosa):
Esta enfermedad lentamente progresiva es más frecuente entre los 30 y 50 años de edad; se caracteriza morfológicamente por la presencia de depósitos subepiteliales que contienen inmunoglobulina a lo largo de la MEG. Al principio de la enfermedad, los glomérulos pueden aparecer normales con microscopia óptica, pero los casos bien desarrollados muestran engrosamiento difuso de la pared capilar. La nefropatía membranosa es idiopática aproximadamente en el 85% de los casos. En el resto (nefropatía membranosa secundaria) esto puede ser secundario a Otras afecciones, incluyendo: 1) infecciones (hepatitis B crónica, sífilis, esquistosomiasis, malaria); 2) tumores malignos, especialmente carcinoma del pulmón y del colon, y melanoma; 3) LES Y otras afecciones autoinmunitarias; 4). exposición a sales inorgánicas (oro, mercurio ), y 5) fármacos (penicilamina, capropril, fármacos antiinflamatorios no esteroideos).
Morfologia: Vista al microscopio óptico con tinción de H y E, el cambio básico en la nefropatía membranosa parece ser un engrosamiento difuso de la MBG. Al microscopio electrónico, este engrosamiento aparente está determinado, en parte, por depósitos subepiteliales que se alojan contra la MBG y están separados uno de otro por pequeñas protrusiones espinosas de la matriz de la MBG, que se forman como reacción a los depósitos (patrón "espícula y cúpula"). Según progresa la enfermedad, estas espículas se cierran sobre los depósitos, incorporándolos en la MBG. Además, los podocitos muestran borrado de los pedicelios. Más adelante, en el curso de la enfermedad, los depósitos incorporados pueden catabolizarse y desaparecer finalmente, dejando cavidades dentro de la MBG. El depósito continuado de la matriz de la membrana basal da lugar a membranas progresivamente más gruesas. Al progresar aún más, los glomérulos pueden esclerosarse. La microscopia con inmunofluorescencia muestra depósitos granulares típicos de inmunoglobulinas y complemento a lo largo de la MBG.

 |
A) Engrosamiento difuso de paredes capilares glomerulares (PAS, 400×). B) Depósitos granulares de IgG delimitando las membranas basales del glomérulo (100×).
|
Glomerulonefritis membranoproliferativa:
La GN membranoproliferativa (GNMP) se manifiesta histológicamente por alteraciones en la MBG y el mesangio, y por proliferación de las células glomerulares. Es responsable del 5 al 10% de los casos de síndrome nefrótico idiopático en niños y en adultos. Algunos individuos presentan solamente hematuria o proteinuria en rangos no nefróticos; otros tienen un cuadro combinado nefrótico-nefrítico. Se reconocen dos tipos principales de GNMP (1 y 2) sobre la base de sus hallazgos ultraestructurales, de inmunofluorescencia, y patogénicos distintos. De los dos tipos, el 1 es, con mucho, el más frecuente (aproximadamente, el 80% de los casos).
Patogenia: Están implicados diferentes mecanismos patogénicos en el desarrollo de la enfermedad tipo 1 y 2. La mayoría de los casos de GNMP tipo 1 parecen estar producidos por inmunocomplejos circulantes, como en la enfermedad del suero crónica, pero se desconoce el antígeno responsable. La GNMP tipo 1 también ocurre en asociación con antigenemia de hepatitis B y C , LES, derivaciones auriculoventriculares infectadas e infecciones extra renales con antigenemia persistente o episódica. La patogenia de la GNMP tipo 2, también conocida como enfermedad por depósitos densos, es menos clara. La anomalía fundamental parece ser una activación excesiva del complemento, que puede estar producida por varios mecanismos
que no implican anticuerpos. Algunos pacientes tienen un autoanticuerpo contra la C3-convertasa, denominado factor nefrítico c3, que se cree que estabiliza la enzima y da lugar a una escisión incontrolada de c3 y activación de la vía alternativa del complemento.
Morfologia: Con microscopia óptica, ambos tipos de GNMP son similares. Los glomérulos son grandes, con apariencia lobular acentuada, y muestran proliferación mesangial y de células endoteliales, así como infiltración de leucocitos. La MBG está engrosada, y la pared de los capilares glomerulares muestra, con frecuencia, un contorno doble o apariencia en "raíles de tranvía", más evidente con tinciones de plata o ácido peryódico de Schiff (PAS). Esto está producido por la escinsión de la MBG debida a la inclusión dentro de la misma de procesos del mesangio y células inflamatorias que se extienden a las asas capilares periféricas.
Los tipos 1 y 2 tienen diferentes características ultraestrueturales y por microscopia con inmunofluorescencia.
la GNMP tipo 1 se caracteriza por depósitos delimitados subendoteliales electrodensos. En la microscopia con inmunofluorescencia, el C3 se deposita en un patrón granular irregular, y también están presentes la IgG y los componentes iniciales del complemento (Clq y C4), lo que indica una patogenia por inmunocomplejos.
En lesiones de tipo 2, la lámina densa y el espacio subendotelial de la MBG se transforman en una estructura irregular, similar a una cinta, extremadamente electro-densa, que resulta del depósito de un material cuya composición se desconoce, dando lugar al término enfermedad por deposito denso.

 |
| A) Proliferación mesangial con incremento de matriz y celularidad mesangial con engrosamiento de paredes capilares (HE, 400×). B) Estudio ultraestructural con depósitos densos en el espacio subendotelial. Inicio de doble contorno. |